
資訊中心
作者:香雪生命科學(xué)研究中心-葉韻
發(fā)布時(shí)間:2021-08-28
閱讀:12282
一、什么是GMP?
GMP是良好生產(chǎn)規范(Good Manufacturing Practice)的英文縮寫(xiě)。全稱(chēng)是Good Manufacturing Practice and Quality Control for Drug(藥品)。當然廣義的GMP還for Food(食品)、for Cosmetic(化妝品)。GMP是指在藥品生產(chǎn)過(guò)程中實(shí)施質(zhì)量管理,保證產(chǎn)品優(yōu)質(zhì)的一套完整的、系統的、科學(xué)的管理規范,是藥品生產(chǎn)和質(zhì)量管理的基本準則。
二、GMP的發(fā)展歷史
GMP的提出和發(fā)展史,是一部人類(lèi)制藥用藥和食品安全的血淚史。
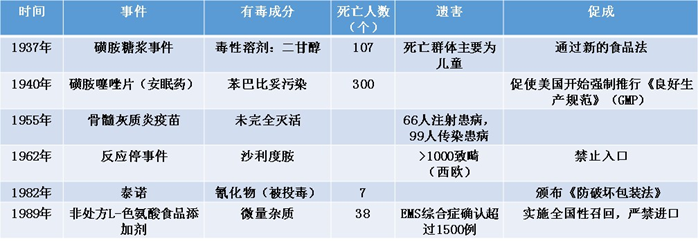
圖一 藥品安全危害事件
GMP的誕生不得不提到FDA(Food and Drug Administration)。自1848年美國聯(lián)邦政府通過(guò)了藥品進(jìn)口法,要求美國海關(guān)總署檢查并阻止劣質(zhì)藥物從海外進(jìn)口,到1906年羅斯福總統簽署《食品藥品法》,人們終于意識到藥品是可以假冒、摻假,食品在不衛生的環(huán)境下生產(chǎn),即便加入防腐劑也是無(wú)效甚至有害的,而這些都需要從法律的途徑去規范。

圖二 美國FDA發(fā)展史
美國國會(huì )于1963年頒布了世界上第一部GMP, 1973年日本制藥工業(yè)協(xié)會(huì )提出了自己的GMP,1974年日本政府頒布GMP,進(jìn)行指導推行。1975年11月 WHO正式公布 GMP。 1980年日本決定正式實(shí)施GMP。
此后,英國、日本及大多數歐洲國家開(kāi)始宣傳、認識、起草本國的GMP,歐洲共同體委員會(huì )頒布了歐共體的 GMP。到1980年有63個(gè)國家頒布了GMP. 目前,已有100多個(gè)國家實(shí)行了GMP制度。 1998年國家藥品監督管理局成立后,建立了國家藥品監督管理局藥品認證管理中心,并于1999年6月18日頒發(fā)了《藥品生產(chǎn)質(zhì)量管理規范(1998年修訂)》。截止2000年年底,我國已有713家藥品生產(chǎn)企業(yè)(車(chē)間)通過(guò)GMP認證。
我國主要的監管機構是CFDA(China Food and Drug Administration),是把食品安全辦的職責、食品藥品監管局的職責、質(zhì)檢總局的生產(chǎn)環(huán)節食品安全監督管理職責、工商總局的流通環(huán)節食品安全監督管理職責整合組建而成,負責藥品、醫療器械、化妝品和消費環(huán)節食品安全的監督管理。2013年3月22日,食品藥品監督管理局改成國家食品藥品監督管理總局,英文簡(jiǎn)稱(chēng)由“SFDA”變成“CFDA”,簡(jiǎn)稱(chēng) “中國食藥監”。
三、GMP的目標
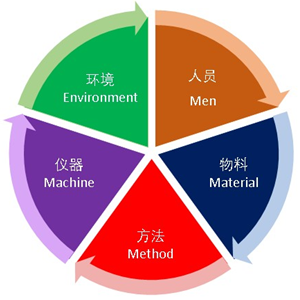
圖三 GMP 5大要素
實(shí)施GMP的主要目標是通過(guò)控制藥品生產(chǎn)過(guò)程中的5大要素,把人為的差錯控制在最低限度,以保證產(chǎn)品的一致性、有效性以及安全性:
(1) 在管理方面
質(zhì)量管理部門(mén)的建立;相互監督檢查制度的建立;制訂規范相應的SOP;整理和保管好記錄;人員的配備、培訓和管理。
(2) 在設備方面
合理的空間布局,不同品種操作必須有相應的時(shí)空分離。
2. 保證產(chǎn)品的安全性
(1) 在管理方面
定期操清掃作室和清潔設備;對生產(chǎn)人員進(jìn)行嚴格的衛生教育;操作人員定期進(jìn)行身體檢查;限制非生產(chǎn)人員進(jìn)入工作間等。
(2) 在設備方面
相應的機械設備(空調凈化系統等)以及定期維護;操作室專(zhuān)用化;對直接接觸藥品的機械設備、工具、容器,選用對藥物不發(fā)生變化的材質(zhì);對無(wú)菌操作區要進(jìn)行微粒檢查和浮游菌、沉降菌的檢查,定期滅菌等。
3.保證高質(zhì)量產(chǎn)品的質(zhì)量管理體系
(1) 在管理方面
定期維修校正設備;檢查生產(chǎn)工序各階段的質(zhì)量、有計劃的合理的質(zhì)量控制、質(zhì)量管理實(shí)施計劃、試驗方案、技術(shù)改造、質(zhì)量攻關(guān)要適應生產(chǎn)計劃要求、追蹤藥品批號,并作好記錄;在適當條件下保存出廠(chǎng)后的產(chǎn)品質(zhì)量檢查留下的樣品、收集消費者對藥品投訴的情報信息,隨時(shí)完善生產(chǎn)管理和質(zhì)量管理等。
(2)在裝備方面
合理配備操作室和機械設備,采用先進(jìn)的設備及合理的工藝布局;為保證質(zhì)量管理的實(shí)施,配備必要的實(shí)驗、檢驗設備和工具等。(2)
四、我們的產(chǎn)品

2018年12月29日,我司具有自主知識產(chǎn)權的高親和性 T 細胞受體(TCR)的抗腫瘤TCR-T 細胞治療新藥----TAEST16001注射液產(chǎn)品獲得國家藥品監督管理局藥品審評中心(CDE,Center for Drug Evaluation)藥品臨床試驗申報(IND, Investigational New Drug Application)申請受理。
參考文獻: